The nervous system is a well-known target for the endocrine effects of hormonal steroids coming from peripheral steroidogenic glands. However, the nervous system is also controlled in a paracrine and autocrine manner by steroids directly synthesized by neurons and glial cells, named neurosteroids. E.E. Baulieu introduced the term ‘‘neurosteroid’’ in 1981 to designate steroids, which are synthesized de novo in the brain via the classic mevalonate pathway to cholesterol and independently of plasma levels. Neuroactive steroids are capable of modifying neural activities by affecting neurotransmitter receptor binding and regulating response to hypothalamo-pituitary-adrenocortical (HPA) axis.
Stress induces a physiological response which is mediated by the HPA axis. CRH is released from the hypothalamus and acts in the pituitary to signal the release of adrenocorticotropic hormone (ACTH), which triggers the release of cortisol from the adrenal gland in humans (corticosterone in mice). The HPA axis is regulated by inputs from numerous different brain regions, involving multiple neurotransmitter systems, as well as the feedback of steroid hormones acting on mineralocorticoid receptors (MRs) and glucocorticoid receptors (GRs) These inputs impinge on CRH neurons in the paraventricular nucleus (PVN), which mediate the output of the HPA axis. Indeed, in severely depressed patients, emotional arousal, cognitive abnormality and vulnerability to psychotic episodes are linked to a hyperactive HPA axis and high level of circulating glucocorticoids, which returns to normal after antidepressant treatment. Although CRH neurons receive a wide variety of inputs from diverse brain regions, their activity is ultimately regulated by GABAergic inhibition. A role for GABA in HPA axis regulation has been well established.
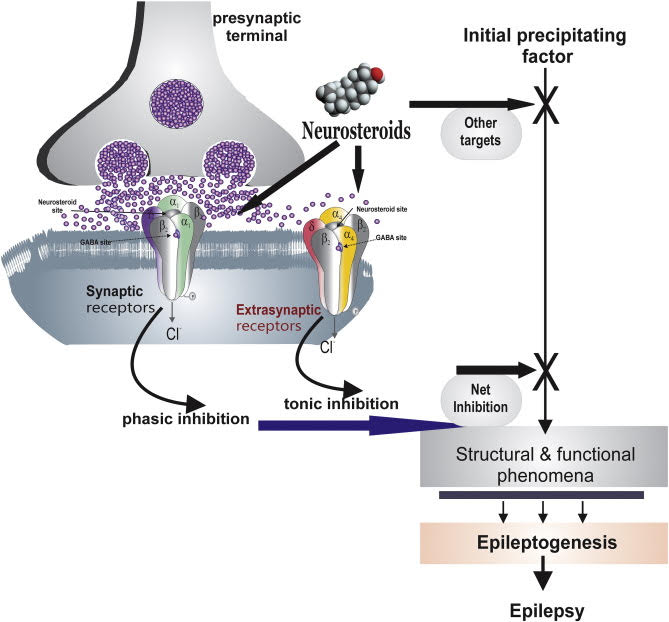
The GABAA receptor is the main target of action of neuroactive steroids. GABAA receptors are hetero pentameric GABA-gated chloride channels: they are involved in fast inhibitory neurotransmission. Composition of the five subunits determines the functional and pharmacological properties of GABAA receptors. The best known and most widely distributed form in the CNS consists of two alphas, two betas, and a third subunit, which together constitute the chloride ion channel. Site-specific binding of the GABAA receptor, in subunit beta, determines the opening of the ion channel and chloride influx. The increase in negative charge leads to a hyperpolarization of the membrane, making it less susceptible to excitation. The activation of GABAA receptors prevents a potential short circuit in the depolarization induced by excitatory neurotransmitters.
The neuroactive steroids increase the flow of chloride ions from GABAA receptors, by increasing both the frequency and duration of the opening of the ion channel. Due to the increased probability of opening of the chloride channel of the GABAA receptor, neuroactive steroids increase a massive influx of the ion and potentiate inhibitory GABAergic transmission. A short, regulatory loop linking neuroactivesteroids’ effects to GABA actions might exist.
Allopregnanolone and other similar neurosteroids act as positive allosteric modulators and direct activators of GABAA receptors. Given the emerging understanding of the role of neurosteroids as autocrine modulators of neuronal excitability, the question arises as to whether endogenous neurosteroids regulate seizure susceptibility. Because in epilepsy inhibitory activity of GABA is inhibited. Lawrence et al. provide important new evidence that the availability of neurosteroids does indeed critically influence the propensity for seizures. These investigators used epileptic female rats that had experienced prolonged bout of status epilepticus induced by lithium– pilocarpine treatment. The epileptic animals exhibited about six seizures per day, each lasting approximately a minute. However, when neurosteroids were withdrawn, using the neurosteroid synthesis inhibitor finasteride, an enormous (more than ten-fold) increase in seizure frequency was observed. In contrast, finasteride—a selective inhibitor of 5α-reductase, the first and rate-limiting enzymatic step in the synthesis of neurosteroids from their steroid hormone precursors (e.g., progesterone)—did not induce seizures in normal animals. Similarly, there is no evidence that finasteride causes seizures in humans who do not have epilepsy.
Lawrence et al. conducted a final experiment in ovariectomized epileptic animals that had low serum progesterone levels maintained by subcutaneous implantation of a progesterone pellet. Unexpectedly, these animals also exhibited a huge (25-fold) increase in seizure frequency following finasteride treatment. Unfortunately, the authors did not study animals in which peripheral progesterone was completely eliminated. Nevertheless, the results of this last experiment suggest that the exacerbation of seizures that occurs following finasteride treatment is due to the inhibition of brain neurosteroid synthesis and is not caused by suppression of the conversion of peripheral (ovarian) progesterone to allopregnanolone. This important conclusion focuses attention squarely on brain neurosteroids as critical regulators of seizures in epilepsy. Clearly, more needs to be learned about the role of locally synthesized neurosteroids in epileptic brain circuits. It can be concluded that potential of neurosteroids may prove as a novel treatment approach for seizures.
Author Madhvi